
10 Clinical Trials to Watch in H2 2026: From Lung Cancer Immunotherapy to Personalised Cancer Vaccines — A Regulatory Affairs Briefing
- 2 days ago
- 12 min read
The most closely watched clinical trials to watch in H2 2026 span lung cancer immunotherapy, personalised mRNA cancer vaccines, obesity drugs, rare neuromuscular disease and novel anticoagulants, according to industry tracking from BioPharma Dive. These trials affect oncologists, biotech investors, sponsors and regulatory affairs teams preparing dossiers across the FDA, EMA and MHRA. Readouts expected between July and December 2026 will shape marketing authorisation strategy, breakthrough therapy designation planning and competitive positioning for several major pharmaceutical companies. This matters because trial design and endpoint choices made now directly determine the regulatory pathway and labelling claims sponsors can pursue later.

Contents
1. At a Glance
At a Glance
- BioPharma Dive identified 10 clinical trials to watch in the second half of 2026, spanning oncology, immunology, cardiology and rare disease.
- The ivonescimab programme from Akeso, Inc. and Summit Therapeutics Inc. — a PD-1/VEGF bispecific antibody — is one of the most closely tracked assets, following its Phase 3 HARMONi-6 results presented at the 2026 ASCO Annual Meeting.
- Moderna, Inc. and Merck & Co., Inc. are progressing the INTerpath Phase 3 programme evaluating personalised neoantigen therapy mRNA-4157 (V940), known by the International Non-proprietary Name intismeran autogene, alongside KEYTRUDA (pembrolizumab).
- Bristol Myers Squibb is testing KarXT (Cobenfy) for Alzheimer's disease psychosis in the ADEPT-2 trial, seeking a label expansion beyond its existing schizophrenia indication.
- Eli Lilly and Company's retatrutide obesity programme (**Triumph trials**) and Novartis's rare neuromuscular disease RNA therapy (**Harbor trial**) both carry significant commercial and regulatory stakes.
- Several trials in this cohort could support Breakthrough Therapy Designation-linked expedited review strategies with the FDA, depending on final readout data.
- Regulatory affairs teams should treat this list as a planning signal, not a confirmed approval timeline — trial completion does not equal submission readiness.
Background: Why H2 2026 Is a Pivotal Window for Oncology and Immunology Trials
The second half of 2026 concentrates an unusually high volume of Phase 3 readouts across oncology and immunology, reflecting a broader industry pattern: sponsors are racing to generate confirmatory survival and efficacy data ahead of a wave of patent expirations affecting major biologics through the late 2020s. This is why personalised cancer vaccines, bispecific antibodies and RNA-based therapies dominate the current watch-list — these modalities represent the next generation of differentiated assets that sponsors hope will anchor their pipelines once legacy blockbusters lose exclusivity. For regulatory affairs teams, this concentration of readouts is significant because each positive Phase 3 result triggers a parallel wave of regulatory activity: pre-submission meetings, breakthrough or PRIME-equivalent designation requests, and dossier preparation across multiple jurisdictions simultaneously.
Interested in building a career in regulatory affairs and working with frameworks like FDA Breakthrough Therapy Designation strategy or EMA PRIME support for oncology and cell-based therapies? The Introduction to Regulatory Affairs course at Entry to Regulatory covers areas relevant to this topic, including Clinical trial application (CTA) and IND regulatory requirements, Breakthrough Therapy Designation and expedited pathway strategy, BLA and MAA dossier structure, oncology and personalised medicine regulatory frameworks, comparative EU/UK/US clinical development regulationIncludes: Up to 3 months real work experience, job mentoring, CV review, mock interviews, industry-recognised certificate Format: Online, part-time | 6 hours per week | 3 months View the course or join our free introductory webinar on Thursday: https://pages.entrytoregulatory.com/courses/
This wave has clear historical roots. The mRNA-4157 (V940) programme, for example, began with Phase 2b data from the KEYNOTE-942 study in high-risk melanoma, which reported a 49% reduction in the risk of recurrence or death at five years when intismeran autogene was combined with pembrolizumab, according to results announced by Merck & Co., Inc. That mid-stage signal was strong enough to earn mRNA-4157 a Breakthrough Therapy Designation from the FDA and to justify launching two large Phase 3 confirmatory trials — INTerpath-001 in resected melanoma and INTerpath-002 in resected non-small cell lung cancer (NSCLC) — both of which are now maturing toward readouts in the H2 2026 window this article covers.
Understanding how a Phase 2b signal translates into a Breakthrough Therapy Designation request and a Phase 3 confirmatory design is a core component of the Entry to Regulatory regulatory affairs training programme, which covers FDA expedited pathway strategy as part of its practical curriculum for EU, UK and US regulations — see the full course details

Lung Cancer Immunotherapy: The Bispecific Antibody Race
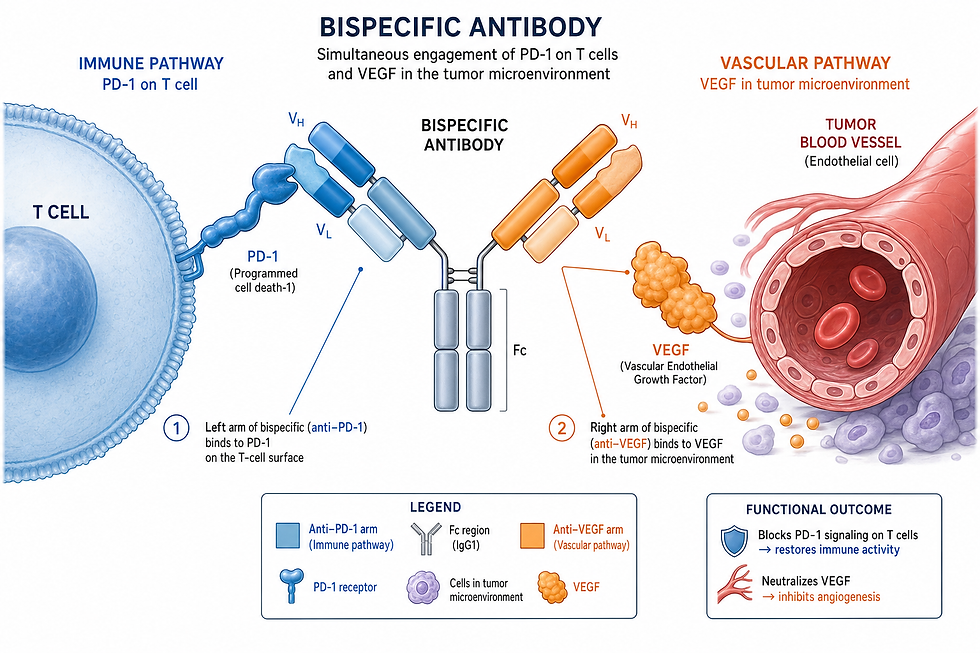
The most closely watched oncology asset in this cohort is ivonescimab, a PD-1/VEGF bispecific antibody co-developed by Akeso, Inc. in China and licensed for development outside Greater China by Summit Therapeutics Inc. Ivonescimab works by simultaneously blocking the PD-1 immune checkpoint and the vascular endothelial growth factor (VEGF) pathway, an approach designed to enhance T-cell activity while also disrupting tumour blood supply.
The programme's most significant recent milestone was the Phase 3 HARMONi-6 trial, conducted by Akeso in China, results from which were presented at the 2026 ASCO Annual Meeting and published in The Lancet. In first-line treatment of advanced squamous non-small cell lung cancer (NSCLC), ivonescimab plus chemotherapy demonstrated:
1. A 34% reduction in the risk of death compared with tislelizumab plus chemotherapy (hazard ratio 0.66).
2. A median overall survival of 27.9 months, versus 23.7 months for the tislelizumab-based control arm.
3. A 24-month overall survival rate of 64.7%, versus 48.6% for the comparator.
> "This outcome is pivotal as it marks the first significant OS benefit in first-line NSCLC compared to PD-1 therapies."
> — Summit Therapeutics Inc., investor announcement, June 2026
Industry commentary from BioPharma Dive has flagged a further, related trial in the ivonescimab programme — internally tracked in some industry coverage as Harmoni-3 — comparing the PD-1/VEGF bispecific approach against standard chemotherapy in NSCLC, as one of the ten trials to monitor through the remainder of 2026. Regulatory affairs professionals should note that the ivonescimab clinical programme spans multiple concurrently running Phase 3 studies (registered separately on ClinicalTrials.gov), and it is important to distinguish which specific trial and comparator arm underlies any given headline before citing efficacy figures in submission-related materials. Regulatory professionals looking to deepen their understanding of how overall survival data from a foreign-conducted pivotal trial supports a global filing strategy will find relevant practical training in the Entry to Regulatory course, which includes hands-on assignments covering multi-region clinical trial data packages as part of its EU, UK and US regulatory curriculum. Full details at https://pages.entrytoregulatory.com/courses/
---
Personalised Cancer Vaccines: mRNA Neoantigen Therapies Move Into Phase 3
The second major theme in this trial cohort is the emergence of personalised, tumour-specific mRNA cancer vaccines as a distinct regulatory category. The lead asset here is mRNA-4157 (V940), also known by the INN intismeran autogene, jointly developed by Moderna, Inc. and Merck & Co., Inc. Unlike a conventional vaccine, intismeran autogene is manufactured individually for each patient based on the specific neoantigen mutation profile of their own tumour, then administered in combination with the anti-PD-1 checkpoint inhibitor KEYTRUDA (pembrolizumab).
Five-year follow-up data from the Phase 2b KEYNOTE-942 study, involving 157 participants with high-risk melanoma, reported:
Outcome (5-year follow-up) | Intismeran autogene + KEYTRUDA | Improvement vs KEYTRUDA alone |
Recurrence-free survival | Sustained improvement | 49% reduction in risk of recurrence or death |
Distant metastasis-free survival | Sustained improvement | 59% reduction in risk |
Overall survival | Sustained improvement | 53% reduction in risk (as reported) |
Building on this signal, Merck and Moderna have initiated two Phase 3 confirmatory trials under the INTerpath programme:
- INTerpath-001 — enrolling approximately 1,089 patients with resected melanoma, evaluating recurrence-free survival, distant metastasis-free survival and overall survival.
- INTerpath-002 — enrolling approximately 868 patients with resected non-small cell lung cancer, using disease-free survival as its primary endpoint.
A related trial, INTerpath-009, is evaluating the same combination specifically in NSCLC patients who did not achieve a pathological complete response following neoadjuvant pembrolizumab and chemotherapy — a patient population regulators and clinicians consider to be at particularly high risk of recurrence. For regulatory affairs professionals, this cluster of trials illustrates a distinctive feature of personalised therapies: the manufacturing and chemistry, manufacturing and controls (CMC) strategy for an individualised product must be validated across an entire trial population before a single marketing authorisation can be sought, since each dose is a unique batch tied to one patient's tumour profile.
Beyond Oncology: Alzheimer's, Rare Disease and Cardiometabolic Trials to Watch
Several trials outside oncology in this cohort carry equally significant regulatory implications:
1. ADEPT-2 (**Bristol Myers Squibb**) — testing KarXT (Cobenfy) for Alzheimer's disease psychosis, seeking to expand the drug's label beyond its existing schizophrenia indication.
2. Embarq trials (**Celldex Therapeutics**) — evaluating barzolvolimab for chronic spontaneous urticaria, targeting a potential breakthrough treatment for a condition with limited existing options.
3. Cardio-TTRansform (**AstraZeneca** and Ionis Pharmaceuticals) — evaluating eplontersen for transthyretin amyloidosis cardiomyopathy.
4. TOPAZ trials (**Biogen**) — assessing litifilimab for systemic lupus erythematosus.
5. Opus-1 (**Vaxcyte, Inc.**) — a new pneumococcal vaccine candidate aiming to outperform existing options.
6. Milvexian trials (**Bristol Myers Squibb** and Johnson & Johnson) — testing a novel oral anticoagulant against current standard-of-care blood thinners.
7. Triumph trials (**Eli Lilly and Company**) — assessing retatrutide for obesity management.
8. Harbor trial (**Novartis**) — investigating an RNA therapy for a rare neuromuscular disease.
Each of these trials sits at a different point on the regulatory spectrum — from label expansion (ADEPT-2) to first-in-class approval strategy (Opus-1, Harbor) — and regulatory affairs teams tracking competitor pipelines should note that a positive readout in any of these categories will likely trigger accelerated designation requests across the FDA, EMA and, where relevant, the MHRA, given the size of the addressable patient populations involved.
Deepen Your Knowledge: Regulatory Affairs Training on This Topic
Tracking a cohort of Phase 3 trials like this one requires regulatory affairs professionals to move fluidly between clinical trial data interpretation, expedited pathway strategy and multi-region submission planning — often for several sponsors' competing assets simultaneously. This is precisely the skill set that separates regulatory affairs professionals who can only summarise a press release from those who can advise a sponsor on what a given readout actually means for their own filing strategy.
The Entry to Regulatory course curriculum addresses these areas directly, with modules covering clinical trial application (CTA) and Investigational New Drug (IND) requirements, Breakthrough Therapy Designation and PRIME-equivalent expedited pathways, and Biologics License Application (BLA) and Marketing Authorisation Application (MAA) dossier structure, alongside comparative coverage of how equivalent oncology and personalised medicine products are reviewed under the EU (EMA), UK (MHRA) and US (FDA) frameworks.
This training is most valuable for life science graduates aiming to enter oncology or immunology-focused regulatory affairs roles, clinical research professionals transitioning into submission-facing regulatory work, and career changers from pharmacy or laboratory science backgrounds who want structured, practical exposure to how real Phase 3 data moves toward regulatory submission.
Practical Implications for Regulatory Affairs Professionals
Sponsors, competitor intelligence teams and regulatory affairs professionals tracking this cohort of trials will have practical questions about how these readouts affect their own strategic planning. The table below addresses the most relevant of these.
Key Question | Previous Situation | What Changes Now |
Is a personalised, patient-specific mRNA vaccine regulated the same way as a conventional biologic? | Limited regulatory precedent existed for reviewing individually manufactured, tumour-specific therapies at Phase 3 scale. | INTerpath-001 and INTerpath-002 will generate the first large-scale precedent for CMC and manufacturing consistency review of a personalised neoantigen therapy. |
Does a foreign-conducted pivotal trial (e.g. HARMONi-6 in China) support a global filing? | Sponsors often needed separate confirmatory trials in Western populations before filing with the FDA or EMA. | Ivonescimab's global Phase 3 programme, including additional Western-population trials, is specifically designed to test whether the China-derived survival benefit generalises for FDA and EMA submissions. |
How does a label-expansion trial like ADEPT-2 differ procedurally from a first-in-class approval? | Label expansions for an already-approved product followed a narrower supplemental application pathway. | A positive ADEPT-2 readout would trigger a supplemental New Drug Application-type submission rather than a full new BLA, significantly shortening the regulatory timeline versus a first-in-class filing. |
What happens if a trial in this cohort misses its primary endpoint? | N/A — endpoint misses historically triggered programme reassessment. | Sponsors will need to decide between programme discontinuation, a redesigned confirmatory trial, or reliance on secondary endpoint data — each carrying different regulatory risk. |
Will positive Phase 3 data automatically translate into Breakthrough Therapy Designation? | Some sponsors assumed strong Phase 2 signals guaranteed expedited designation continuity into Phase 3. | Breakthrough Therapy Designation granted at Phase 2 does not guarantee automatic acceleration through Phase 3; sponsors must demonstrate continued substantial improvement over available therapies. |
How should competitor intelligence teams track multiple concurrently running trials within one drug programme? | Ambiguity between similarly named trials (e.g. HARMONi-6 versus other HARMONi-numbered studies) has caused sourcing errors in industry reporting. | Regulatory and competitive intelligence teams should verify exact trial registration numbers via ClinicalTrials.gov before citing efficacy data in internal or external materials. |
Key Takeaways
1. Verify exact trial identifiers before citing efficacy data from multi-trial programmes such as ivonescimab's HARMONi series, given the risk of conflating results from different comparator arms.
2. Track Breakthrough Therapy Designation status separately from trial phase — designation granted at Phase 2 does not carry an automatic guarantee through to Phase 3 outcomes.
3. Prepare CMC strategy early if you are developing or advising on a personalised, patient-specific therapy, since manufacturing consistency review for individualised products differs substantially from conventional biologics.
4. Map competitor label-expansion trials like ADEPT-2 against your own portfolio to anticipate supplemental application activity in adjacent therapeutic areas.
5. Monitor ASCO, ESMO and equivalent conference disclosures through H2 2026, as several of these trials are expected to report topline or detailed data at major oncology congresses.
6. Distinguish overall survival from progression-free or recurrence-free survival claims precisely when briefing internal stakeholders, given regulators' differing weight on each endpoint type.
7. Benchmark your own pipeline's competitive position against this cohort, since multiple readouts in the same therapeutic area (e.g. NSCLC) within a single half-year window will materially affect market access and pricing negotiations.
Take the Next Step in Your Regulatory Affairs Career
Following a cohort of trials like this one — across oncology, immunology and rare disease, spanning bispecific antibodies, personalised mRNA vaccines and RNA therapies — is exactly the kind of work regulatory affairs professionals do every day. Understanding how a Phase 3 readout translates into a designation request, a dossier strategy or a competitive threat is a core skill at every stage of a regulatory affairs career, from entry-level roles through to senior strategic positions.
If this kind of analysis interests you, the Introduction to Regulatory Affairs course at Entry to Regulatory is a practical next step, covering the FDA, EMA and MHRA frameworks that govern trials like these. A free introductory webinar is available if you would like to explore the course with no commitment.
Explore the full course details and register for a free introductory webinar at Entry to Regulatory: https://pages.entrytoregulatory.com/courses/

---
Frequently Asked Questions
### What is the most significant lung cancer trial to watch in H2 2026?
The ivonescimab programme from Akeso, Inc. and Summit Therapeutics Inc. is among the most closely watched, following Phase 3 HARMONi-6 results showing a 34% reduction in risk of death versus tislelizumab plus chemotherapy in advanced squamous non-small cell lung cancer, presented at the 2026 ASCO Annual Meeting.
### What is a personalised cancer vaccine and how is it regulated differently from a standard vaccine?
A personalised cancer vaccine, such as intismeran autogene (mRNA-4157/V940), is manufactured individually for each patient based on their tumour's specific mutation profile, meaning regulators must review manufacturing consistency and quality control processes designed for individualised, small-batch production rather than a single standardised product.
### Why does Breakthrough Therapy Designation matter for these trials?
Breakthrough Therapy Designation allows the FDA to work more closely with a sponsor during development and can support a faster review timeline, but it does not guarantee that Phase 3 data will confirm the Phase 2 signal that earned the designation in the first place.
### How many trials are on this H2 2026 watch-list and what areas do they cover?
BioPharma Dive identified 10 trials to watch in the second half of 2026, spanning lung cancer immunotherapy, personalised melanoma and lung cancer vaccines, Alzheimer's disease psychosis, chronic spontaneous urticaria, cardiac amyloidosis, lupus, pneumococcal vaccination, anticoagulation, obesity and rare neuromuscular disease.
### How can I build the regulatory affairs skills needed to track and act on trials like these?
Understanding how Phase 3 oncology and immunology trial data feeds into designation strategy and dossier preparation is a core regulatory affairs skill, and the Introduction to Regulatory Affairs course at Entry to Regulatory covers this directly across the US, EU and UK, alongside practical work experience and job search mentoring — see https://pages.entrytoregulatory.com/courses/
Next cohort starts: 13 August 2026. Limited spaces available.
Further Reading and Reference Sources
1. 10 Clinical Trials to Watch in the Second Half of 2026 — BioPharma Dive (https://www.biopharmadive.com/news/biotech-pharma-clinical-trials-watch-2026/808255/)
2. Akeso, Summit Drug Extends Survival in Closely Watched Lung Cancer Trial — BioPharma Dive (https://www.biopharmadive.com/news/akeso-summit-ivonescimab-asco-2026-harmoni6/821559/)
3. HARMONi-6 Demonstrates Significant Overall Survival Benefit — Akeso, Inc. (https://www.akesobio.com/en/media/akeso-news/260531/)
4. Ivonescimab with Chemotherapy Demonstrated a Statistically Significant Overall Survival Benefit — Summit Therapeutics Inc. (https://smmttx.com/news/press-releases/news-details/2026/Ivonescimab-with-Chemotherapy-Demonstrated-a-Statistically-Significant-Overall-Survival-Benefit-Compared-to-Tislelizumab-Plus-Chemotherapy-in-1L-Treatment-of-Patients-with-Squamous-NSCLC-in-the-HARMONi-6-Study-Conducted-by-Akeso-in-China/default.aspx) — Published: June 2026
5. Moderna & Merck Announce 5-Year Data for Intismeran Autogene in Combination with KEYTRUDA — Merck & Co., Inc. (https://www.merck.com/news/moderna-merck-announce-5-year-data-for-intismeran-autogene-in-combination-with-keytruda-pembrolizumab-demonstrated-sustained-improvement-in-the-primary-endpoint-of-recurrence-free-survival-i/)
6. Merck and Moderna Initiate INTerpath-002, a Phase 3 Study — Merck & Co., Inc. (https://www.merck.com/news/merck-and-moderna-initiate-interpath-002-a-phase-3-study-evaluating-v940-mrna-4157-in-combination-with-keytruda-pembrolizumab-for-adjuvant-treatment-of-patients-with-certain-types-of-resect/)
7. A Hotly Debated Lung Cancer Drug Cut the Risk of Death by 34% in a Late-Stage Trial in China — CNBC (https://www.cnbc.com/2026/05/31/asco-summit-akeso-ivonescimab-improves-survival-in-harmoni-6-trial.html)
8. ASCO: Moderna's mRNA-Based Melanoma Vaccine Shows 'Encouraging' 5-Year Survival — BioSpace (https://www.biospace.com/drug-development/asco-modernas-mrna-based-melanoma-vaccine-shows-encouraging-5-year-survival)
About the Author: Rabiea is an Honorary Associate Professor at UCL, former MHRA Health Authority reviewer, and CEO of Entry to Regulatory and Advanced Regulatory Consulting. After transitioning from retail pharmacy to regulatory affairs, she has dedicated her career to helping others make the same successful career change. Connect with her on LinkedIn for the latest regulatory affairs insights and career advice.
Disclaimer
This article is provided for informational purposes only. Regulatory guidance, legislative instruments and health authority policies evolve frequently. Always consult the most current official publications from the relevant health authority — including the MHRA, UK Space Agency and Civil Aviation Authority — and seek qualified professional regulatory advice for specific product development, submission or compliance decisions. Entry to Regulatory training courses are designed for educational and career development purposes.



Comments